
Liegt der Verdacht einer Demenzerkrankung nahe, suchen Betroffene und deren Angehörige zunächst meist den Rat eines Hausarztes. Der Ablauf der Erstellung einer Demenz-Diagnose beim Arzt gestaltet sich wie folgt:
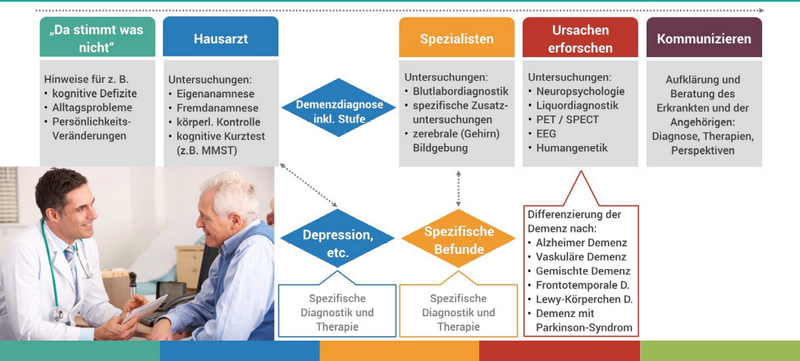
Die in der Abbildung 2 aufgeführten neuropsychologischen kognitiven Kurztests wie Mini Mental State Test (MMST), ADAS-COG (Standard, hauptsächliche Verwendung im anglo-amerikanischen Raum), Montreal Cognitive Assessment (MoCA), ebenso DemTect (Ruhr-Universität Bochum) liefern bei positivem Befund zunächst ein ggf. starkes Indiz für das Vorhandensein einer Demenz-Erkrankung.
Die Sensitivität dieser Verfahren bei leichtgradiger und fraglicher Demenz ist meist jedoch aufgrund ihrer groben Granularität und ihres statischen Aufbaus begrenzt. Sie sind nicht zur Differenzialdiagnostik verschiedener Demenzen geeignet, insbesondere nicht der AD mit zwei Drittel aller Demenz-Fälle.
Das wesentliche Problem aller neuropsychologischen Testverfahren besteht im Allgemeinen darin, dass sie nur den kognitiven Ist-Zustand mehr oder weniger genau anzeigen. In einer präklinischen Phase der Entwicklung der Alzheimer-Krankheit sind aber noch keine kognitiven Verfalls-Symptome auffällig.
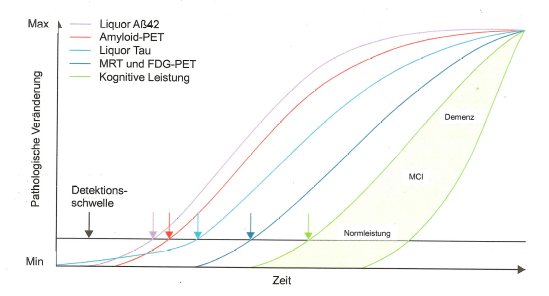
Wie in Abbildung 3 zu erkennen ist, verändern sich die diagnostischen Bio-Marker bereits lange, bevor Symptome der kognitiven Degradation auftreten.
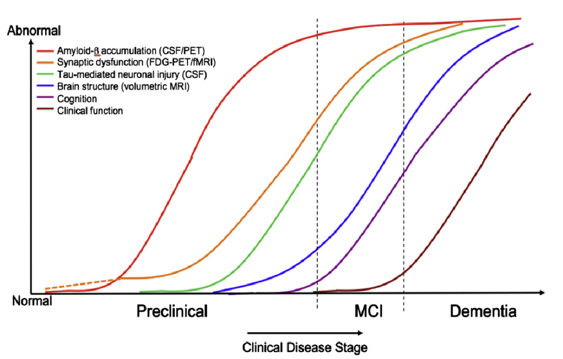
In der nachfolgenden Abbildung 4 sind die Phasen präklinisch, MCI (mild cognitive impairment) und Demenz in Abhängigkeit der Entwicklung der Biomarker angegeben.
Um die Beschränkungen neuropsychologischer Tests zur Feststellung des Schweregrads einer Alzheimer-Erkrankung oder deren Vorläufer zu vermeiden, werden in der Regel bildgebende Verfahren wie Magnetresonanztomographie (MRT) oder Positronen-Emissions-Tomographie (PET-FDG “Fluor-Desoxyglukose“, d.h. schwach radioaktiv markierter Zucker) zur Abgrenzung von AD von anderen Demenz-Formen eingesetzt. MRT und vor allem PET sind jedoch teuer und erfordern einen erheblichen personellen Aufwand mit entsprechendem Expertenwissen. Zudem ist bei diesen Verfahren die Verwendung von Kontrastmitteln unabdingbar, über deren Gesundheitsrisiken (insbesondere bei Gadolinium) die Meinung in den letzten Jahren umschwenkte. Der Einsatz von Labordiagnostik zur Analyse der Hirnflüssigkeit (Liquor) und des Blutes auf Rückstände von Amyloid-β-Plaques ist noch im Stadium der Grundlagenforschung.[3]
In den Abb. 3 und 4 werden die Biomarker Amyloid-β (-42, d.h. die Peptid-Kettenlänge beträgt 42, siehe https://de.wikipedia.org/wiki/Beta-Amyloid) und Tau (p-Tau, d.h. in seiner hyperphosphorylierten Form, siehe https://de.wikipedia.org/wiki/Tau-Protein) als maßgebliche messbare Indikatoren des Vorhandenseins und der Entwicklung des Schweregrads der Alzheimer-Demenz (AD) sowie deren Vorläufer MCI gezeigt.
Bis vor kurzem wurde primär das Vorhandensein präseniler Plaques (Verklumpungen von Amyloid-β, vornehmlich der Kettenlänge 42) im Zwischenraum der Nervenzellen als klares Anzeichen des Vorliegens einer Demenz vom Typ Alzheimer angesehen. Dies ist die Grundlage der „Amyloid-Kaskaden-Hypothese“, die jahrelang die Entwicklung der Forschung nach Medikamenten bestimmte, welche in der Lage sein sollten, die Amyloid-β-Plaques aufzulösen und den Gedächtnisverlust aufzuhalten bzw. gar rückgängig zu machen. Allerdings waren hierbei auch erhebliche Rückschläge zu verzeichnen, siehe z.B. https://www.faz.net/aktuell/wissen/medizin-ernaehrung/ein-schmerzhafter-flop-fuer-die-alzheimer-forschung-14541369.html.
Mittlerweile ist man aufgrund umfangreicher Forschungen davon abgerückt, dass alleine Amyloid-β für die Entwicklung der Alzheimer-Krankheit und des damit einhergehenden Gedächtnisverlusts verantwortlich ist bzw. sein soll. Dass dies nicht der Fall ist, ist in den Abbildungen 3 und 4 gut erkennbar. Prinzipiell gilt: es müssen immer beide Proteine, d.h. Amyloid-β und p-Tau, in ausgeprägter Form und Menge vorhanden sein, damit eine AD mit erkennbaren Symptomen (d.h. festzustellen über neuropsychologische Tests) manifest wird.
Interpretation von Abb. 3 und 4:
Amyloid-β und p-Tau können nach Lumbalpunktion im Hirnwasser gemessen und vor allem mittels FDG-PET (aber auch über MRT) in ihrer Wirkung auf den Glukosestoffwechsel und Blutkreislauf im Gehirn bestimmt werden. Hierdurch können dann über statistische Verfahren die Regionen im Gehirn angegeben werden, die durch die Alzheimer-Demenz zum Untersuchungszeitpunkt besonders betroffen sind. Die Messungen der beiden Biomarker-Proteine im Hirnwasser stellen eine gute Ergänzung zu den bildgebenden Verfahren dar, welche die differentialdiagnostische Qualität erhöhen. So nimmt Amyloid-β (1-42) bei einer manifesten AD im Hirnwasser ab und Tau nimmt zu, siehe z.B. https://www.labor-limbach.de/fuer-aerzte/alle-informationen/abklaerung-einer-alzheimerdemenz/.
Amyloid-β wird immer produziert, bei jeder neuronalen Aktivität. Es wird normalerweise im NREM-Schlaf während der Aktivität des glymphatischen Systems[4] abgebaut.
D.h., es findet ein beständiger Auf- und Abbau der Amyloid-β-Menge im extrazellulären Raum des Gehirns statt. Es handelt sich hierbei aber primär um lösliche Amyloid-β-Peptid-Ketten. Wenn es jedoch u.a. aufgrund der Lebensführung zu Störungen dieses dynamischen Gleichgewichts des Amyloid-β-Auf- und -Abbaus kommt, z.B. durch kontinuierlich gestörten Schlaf, Schlafmangel oder Schlafentzug, dann steigt mit der zunehmenden Amyloid-β-Menge auch die Wahrscheinlichkeit des Auftretens längerkettiger Amyloid-β-Moleküle, die sich zu unlöslichen Plaques verklumpen.
In der Entwicklung der Alzheimer-Pathogenese tritt ab einem bestimmten Zeitpunkt (einem Kipp-Punkt) ein zweites Protein, das Tau-Protein, in veränderter Form auf. Es ist hyperphosphoryliert und verliert hierdurch die Fähigkeit, das Zellgerüst (die Mikrotubuli, über die die Zelle u.a. mit Nährstoffen versorgt wird) zu stabilisieren. Aus diesem Grund bricht das Zellgerüst zusammen und die Nervenzelle stirbt. Dieser Prozess setzt sich, beginnend beim Hippocampus und dem entorhinalen Cortex, wie eine Infektion von einer Nervenzelle zu einer größeren Zahl verbundener Nervenzellen fort, so dass nach wenigen Jahren die ersten Gedächtnisverluste, erst als MCI, später dann auch mit manifester Alzheimer-Symptomatik, festgestellt werden können.
Im November 2019 wurden von Ising, Heneka et al. Ergebnisse ihrer Forschungen veröffentlicht, welche aufzeigen, dass die Hyperphosphorylierung des Tau-Proteins, die den Beginn der eigentlichen Alzheimer-Pathogenese markiert, ab einer bestimmten (individuell verschiedenen) Amyloid-β-Menge aufgrund der Aktivierung eines weiteren Protein-Komplexes, eines Inflammasoms, in die Wege geleitet wird, siehe hierzu auch unter dem Menüpunkt „Aktualisierungen zum Stand der Wissenschaft“ die entsprechenden Ausführungen unter „NLRP3 Inflammasom - ein molekularer Schalter“.
Wenn sich also – aus welchen Gründen auch immer – eine bestimmte Menge von Amyloid-β-Plaque wie auch weiteres, nicht verklumptes Amyloid-β gebildet hat, dann wird dies vom hirneigenen Immunsystem als Entzündung interpretiert und die von der Amyloid-β-Plaque betroffenen Nervenzellen durch Zerstörung ihres Zellgerüsts (Hyperphosphorylierung ⇒ p-Tau) zum Sterben veranlasst. Hierbei bilden sich die typischen Tau-Fibrillen, die das hirneigene Immunsystem ihrerseits zu weiteren entsprechenden Reaktionen veranlassen. Im Menüpunkt „Stand der Technik“ und unter „Methoden“ wird auf die Konsequenzen dieser Erkenntnisse für das IASON-Projekt noch näher eingegangen.
Es wurde aber auch eine Reihe von alternativ möglichen Ursachen der AD angegeben, wie z.B. das chronische Eindringen von Bakterien des Zahnraums[5] in das Gehirn oder durch chronische Schlafstörungen verursachte Behinderungen des glymphatischen Systems[4] in seiner Abtransport-Funktion, die z.B. die Störung der Synchronizität der neuronalen Aktivitäten in den verschiedenen Hirnarealen verursachen können. Die Aufrechterhaltung einer sehr fein abgestimmten Synchronisierung der Aktivitäten miteinander verbundener Hirnregionen (zum Teil durch Interneuronen mit elektrischen Synapsen) ist wesentlich für die Gedächtniskonsolidierung[6], die de facto nur im Schlaf stattfindet. Weitere neueste alternative Erklärungen zur Ursache von AD beziehen sich insbesondere auf die Funktion der Chaperone[7] und deren Unterstützung einer korrekten Proteinfaltung[8].
- ↑ H. Dohmeyer, „Demenz-Diagnose-Prozess“ (2016) https://denken.de/demenz-diagnose-prozess-infografik/demenz-diagnose-prozess-infografik/
- ↑ a b Entnommen aus F. Jessen, „S3-Leitline „Demenzen“, PDF, S.16“, Uniklinik Köln, DGN (2016)
- ↑ T. Müller, „Endlich ein Bluttest auf Alzheimer?“ (2018), https://www.aerztezeitung.de/medizin/krankheiten/demenz/article/957110/demenz-diagnostik-bluttest-alzheimer-amyloid-serum.html
- ↑ a b "Glymphatisches System" in Wikipedia: https://de.wikipedia.org/wiki/Glymphatisches_System
- ↑ S. Dominy et al, „Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors” (2019), Sci. Adv. 2019; 5: 23 January 2019
- ↑ S. Diekelmann & J. Born, "The memory function of sleep", nature reviews neuroscience 11, 114-126 (2010) https://www.researchgate.net/publication/40834254_Diekelmann_S_Born_J_The_memory_function_of_sleep_Nat_Rev_Neurosci_11_114-126
- ↑ "Chaperon (Protein)" in Wikipedia: https://de.wikipedia.org/wiki/Chaperon_(Protein)
- ↑ P. Leiner, "Proteinverklumpung in Neuronen im Visier" (2019) https://www.aerztezeitung.de//panorama/gesellschaft/article/983461/nachwuchspreistraegerin-dr-dormann-gespraech-proteinverklumpung-neuronen-visier.html
 Deutsch
Deutsch  English
English