
If dementia is suspected, patients and their relatives usually seek the advice of a general practitioner first. The procedure for making a dementia diagnosis by a doctor is as follows:
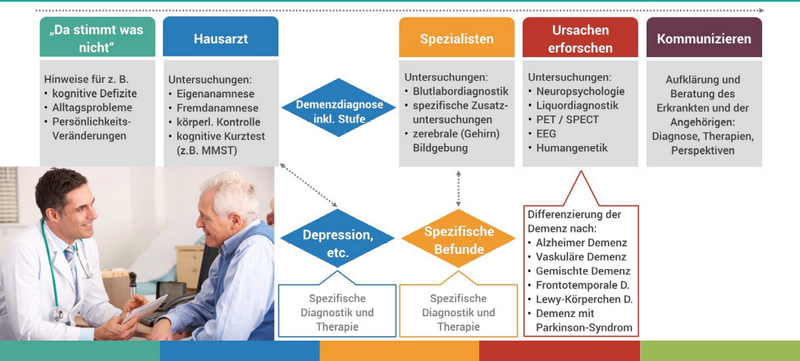
The neuropsychological cognitive short tests listed in Figure 2, such as the Mini Mental State Test (MMST), ADAS-COG (standard, mainly used in the Anglo-American region), Montreal Cognitive Assessment (MoCA), as well as DemTect (Ruhr University Bochum), provide a potentially strong indication of the presence of dementia if the results are positive.
However, the sensitivity of these procedures for mild and questionable dementia is usually limited due to their coarse granularity and static structure. They are not suitable for the differential diagnosis of various dementias, especially AD with two thirds of all dementia cases.
The main problem of all neuropsychological test procedures is generally that they only indicate the cognitive actual state more or less accurately. In a preclinical phase of the development of Alzheimer's disease, however, no cognitive symptoms of decay are noticeable.
As can be seen in Figure 3, diagnostic bio-markers change long before symptoms of cognitive degradation appear.
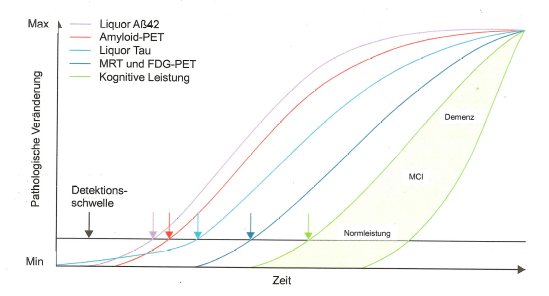
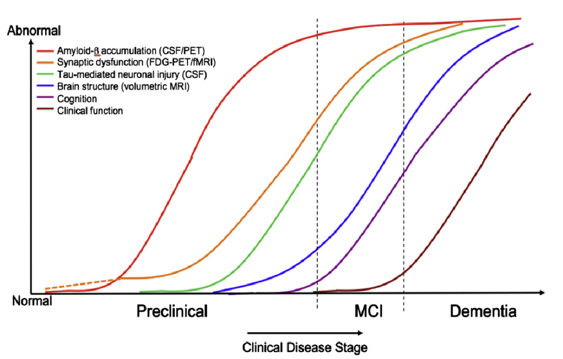
Figure 4 below shows the phases preclinical, MCI (mild cognitive impairment) and dementia depending on the development of the biomarkers.
In order to avoid the limitations of neuropsychological tests to determine the severity of Alzheimer's disease or its precursors, imaging techniques such as magnetic resonance imaging (MRI) or positron emission tomography (PET-FDG "fluorodeoxyglucose", i.e. low-level radioactively labelled sugar) are generally used to distinguish AD from other forms of dementia. However, MRI and especially PET are expensive and require a considerable amount of personnel with appropriate expert knowledge. In addition, these procedures require the use of contrast media, the health risks of which (especially gadolinium) have been the subject of a change of opinion in recent years. The use of laboratory diagnostics for the analysis of cerebrospinal fluid (liquor) and blood for residues of amyloid-β plaques is still in the stage of basic research.[3]
Figures 3 and 4 show the biomarkers amyloid-β (-42, i.e. the peptide chain length is 42, see https://en.wikipedia.org/wiki/Amyloid_beta) and tau (p-tau, i.e. in its hyperphosphorylated form, see https://en.wikipedia.org/wiki/Tau_protein) as key measurable indicators of the presence and development of the severity of Alzheimer's dementia (AD) and its precursor MCI.
Until recently, the presence of presenile plaques (clumps of amyloid-β, mainly of chain length 42) in the space between the nerve cells was considered a clear sign of Alzheimer's dementia. This is the basis of the "amyloid cascade hypothesis", which for years determined the development of research into drugs that should be able to dissolve the amyloid-β plaques and halt or even reverse the memory loss. However, there were also considerable setbacks, see e.g. https://www.faz.net/aktuell/wissen/medizin-ernaehrung/ein-schmerzhafter-flop-fuer-die-alzheimer-forschung-14541369.html.
In the meantime, based on extensive research, there has been a shift away from the view that amyloid-β alone is or should be responsible for the development of Alzheimer's disease and the associated loss of memory. That this is not the case can be clearly seen in figures 3 and 4. In principle, both proteins, i.e. amyloid-β and p-tau, must always be present in pronounced form and quantity for AD to manifest itself with recognisable symptoms (i.e. determined by neuropsychological tests).
Interpretation of figures 3 and 4:
Amyloid-β and p-tau can be measured in the cerebral fluid after lumbar puncture and their effect on glucose metabolism and blood circulation in the brain can be determined mainly by means of FDG-PET (but also by MRI). Statistical methods can then be used to identify the regions of the brain that are particularly affected by Alzheimer's dementia at the time of the examination. The measurements of the two biomarker proteins in the cerebral fluid are a good complement to the imaging procedures, which increase the differential diagnostic quality. For example, amyloid-β (1-42) decreases in the cerebral fluid in manifest AD and tau increases, see for example https://www.labor-limbach.de/fuer-aerzte/alle-informationen/abklaerung-einer-alzheimerdemenz/.
Amyloid-β is always produced during every neuronal activity. It is normally broken down in NREM sleep during activity of the glymphatic system.[4]
This means that a constant build-up and breakdown of the amyloid-β quantity takes place in the extracellular space of the brain. However, these are primarily soluble amyloid-β peptide chains. However, if, for example due to lifestyle, disturbances of this dynamic balance of amyloid-β build-up and degradation occur, e.g. due to continuously disturbed sleep, sleep deficit or sleep deprivation, then the probability of the occurrence of longer-chained amyloid-β molecules that clump together to form insoluble plaques increases with the increasing amyloid-β quantity.
In the development of Alzheimer's pathogenesis, a second protein, the Tau protein, appears in altered form from a certain point in time (a tipping point). It is hyperphosphorylated and thus loses the ability to stabilize the cytoskeleton (the microtubules through which the cell is supplied with nutrients, among other things). For this reason, the cytoskeleton breaks down and the nerve cell dies. This process continues, starting with the hippocampus and the entorhinal cortex, like an infection from one nerve cell to a larger number of connected nerve cells, so that after a few years the first memory losses, first as MCI, and later with manifest Alzheimer symptoms, can be detected.
In November 2019, Ising, Heneka et al. published results of their research which show that hyperphosphorylation of the tau protein, which marks the beginning of the actual pathogenesis of Alzheimer's, is initiated from a certain (individually different) amount of amyloid-β due to the activation of another protein complex, an inflammasome; see also the corresponding explanations under "NLRP3 Inflammasome - a molecular switch" under the menu item "Updates on the state of the art".
Therefore, if – for whatever reason – a certain amount of amyloid-β plaque as well as other non-clumped amyloid-β has formed, then this is interpreted as an inflammation by the brain's own immune system and the nerve cells affected by the amyloid-β plaque are caused to die by destruction of their cytoskeleton (hyperphosphorylation ⇒ p-Tau). This leads to the formation of the typical tau fibrils, which in turn cause the brain's own immune system to react further. In the menu item "state of the art" and under "Methods" the consequences of these findings for the IASON project are discussed in more detail.
However, a number of alternative possible causes of AD have also been suggested, such as chronic invasion of bacteria of the dental space[5] into the brain or obstructions of the glymphatic system[4] in its evacuation function caused by chronic sleep disorders, which can cause, for example, the disruption of the synchronicity of neuronal activities in the different areas of the brain. The maintenance of a very finely tuned synchronization of the activities of interconnected brain regions (partly by interneurons with electrical synapses) is essential for memory consolidation[6], which de facto only takes place during sleep. Other recent alternative explanations of the cause of AD relate in particular to the function of chaperones[7] and their support of correct protein folding[8].
- ↑ H. Dohmeyer, „Demenz-Diagnose-Prozess“ (2016) https://denken.de/demenz-diagnose-prozess-infografik/demenz-diagnose-prozess-infografik/
- ↑ a b Entnommen aus F. Jessen, „S3-Leitline „Demenzen“, PDF, S.16“, Uniklinik Köln, DGN (2016)
- ↑ T. Müller, „Endlich ein Bluttest auf Alzheimer?“ (2018), https://www.aerztezeitung.de/medizin/krankheiten/demenz/article/957110/demenz-diagnostik-bluttest-alzheimer-amyloid-serum.html
- ↑ a b "Glymphatisches System" in Wikipedia: https://de.wikipedia.org/wiki/Glymphatisches_System
- ↑ S. Dominy et al, „Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors” (2019), Sci. Adv. 2019; 5: 23 January 2019
- ↑ S. Diekelmann & J. Born, "The memory function of sleep", nature reviews neuroscience 11, 114-126 (2010) https://www.researchgate.net/publication/40834254_Diekelmann_S_Born_J_The_memory_function_of_sleep_Nat_Rev_Neurosci_11_114-126
- ↑ "Chaperon (Protein)" in Wikipedia: https://de.wikipedia.org/wiki/Chaperon_(Protein)
- ↑ P. Leiner, "Proteinverklumpung in Neuronen im Visier" (2019) https://www.aerztezeitung.de//panorama/gesellschaft/article/983461/nachwuchspreistraegerin-dr-dormann-gespraech-proteinverklumpung-neuronen-visier.html
 English
English  Deutsch
Deutsch